from typing import Union, List
from Bio import AlignSmith-Waterman Local Alignment using Python
bioinformatics
alignment
smith-waterman
biopython
Smith-Waterman is a local alignment method for sequence alignment. Below is example implementation using python.
Note
This post was written using:
- biopython: 1.78
def get_score(A:str, B:str, mismatch_penalty:int, match_score:int) -> int:
# match
if A == B:
return match_score
# mismatch
return mismatch_penalty
def init_matrix(A: str, B: str) -> list:
lenA = len(A) + 1
lenB = len(B) + 1
matrix = []
for i in range(lenB):
matrix.append([0] * lenA)
return matrix
def noNeg(x:int) -> int:
return max(0, x)
def SmithWaterman(A, B, gap_penalty:int=-2, mismatch_penalty:int=-1, match_score:int=4) -> Union[list, int, list]:
""" initialize matrix and fill
Returns:
list: 2D array of filled value according to Smith-Waterman algorithm
int: Max value in the final filled `matrix`
list: List of position [row, col] of `max_score` in `matrix`
"""
matrix = init_matrix(A, B)
# in sw, lower bound to 0
for m in range(len(matrix)):
for n in range(len(matrix[0])):
matrix[m][n] = noNeg(matrix[m][n])
diag = [[-1, -1]]
top = [[-1, 0]]
left = [[0, -1]]
max_score = 0
max_score_position = []
for row in range(1, len(B)+1):
for col in range(1, len(A)+1):
a_char = A[col-1]
b_char = B[row-1]
for dr,dc in left:
l = matrix[row + dr][col + dc] + gap_penalty
for dr,dc in top:
t = matrix[row + dr][col + dc] + gap_penalty
for dr,dc in diag:
d = matrix[row + dr][col + dc] + get_score(a_char, b_char, mismatch_penalty, match_score)
# l,t,d lower bouded to 0 (SW property)
cur_score = max(noNeg(l), noNeg(t), noNeg(d))
if cur_score > max_score:
max_score = cur_score
max_score_position = [row, col]
matrix[row][col] = cur_score
return matrix, max_score, max_score_positiondef traceback(matrix: list, A: str, B: str,
max_score_position: list,
gap_penalty: int,
mismatch_penalty: int,
match_score: int) -> List[str]:
aligned_A = []
aligned_B = []
row = max_score_position[0]
col = max_score_position[1]
while row > 0 and col > 0:
d = matrix[row - 1][col - 1]
t = matrix[row - 1][col]
l = matrix[row][col - 1]
# Stop when we reach a score of 0 (SW property)
if matrix[row][col] == 0:
break
# Diagonal move (match/mismatch)
if matrix[row][col] == d + get_score(A[col-1], B[row-1], mismatch_penalty, match_score):
aligned_A.append(A[col-1])
aligned_B.append(B[row-1])
row -= 1
col -= 1
# Left move (gap in B)
elif matrix[row][col] == l + gap_penalty:
aligned_A.append(A[col-1])
aligned_B.append('-')
col -= 1
# Top move (gap in A)
elif matrix[row][col] == t + gap_penalty:
aligned_A.append('-')
aligned_B.append(B[row-1])
row -= 1
return ''.join(reversed(aligned_A)), ''.join(reversed(aligned_B))## Params
GAP_PENALTY = -2
MISMATCH_PENALTY = -1
MATCH_SCORE = 2
A, B = "AACG", "AATCG" # A = top, B = left
matrix, max_score, max_score_position = SmithWaterman(A, B,
gap_penalty = GAP_PENALTY,
mismatch_penalty = MISMATCH_PENALTY,
match_score = MATCH_SCORE)
for m in matrix:
print(m)[0, 0, 0, 0, 0]
[0, 2, 2, 0, 0]
[0, 2, 4, 2, 0]
[0, 0, 2, 3, 1]
[0, 0, 0, 4, 2]
[0, 0, 0, 2, 6]max_score6max_score_position[5, 4]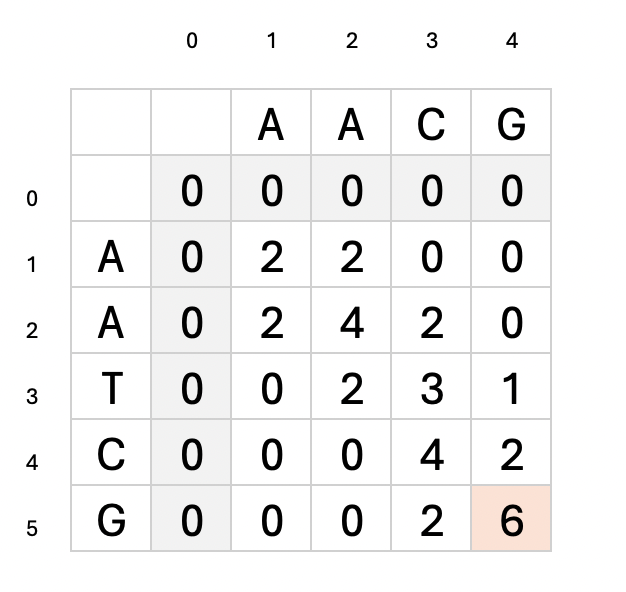
Above is the filled in matrix. The highlighted cell is the max_score.
Traceback
aligned_A, aligned_B = traceback(matrix, A, B,
max_score_position,
gap_penalty = GAP_PENALTY,
mismatch_penalty = MISMATCH_PENALTY,
match_score = MATCH_SCORE)
print("Alignment:")
print(aligned_A)
print(aligned_B)Alignment:
AA-CG
AATCG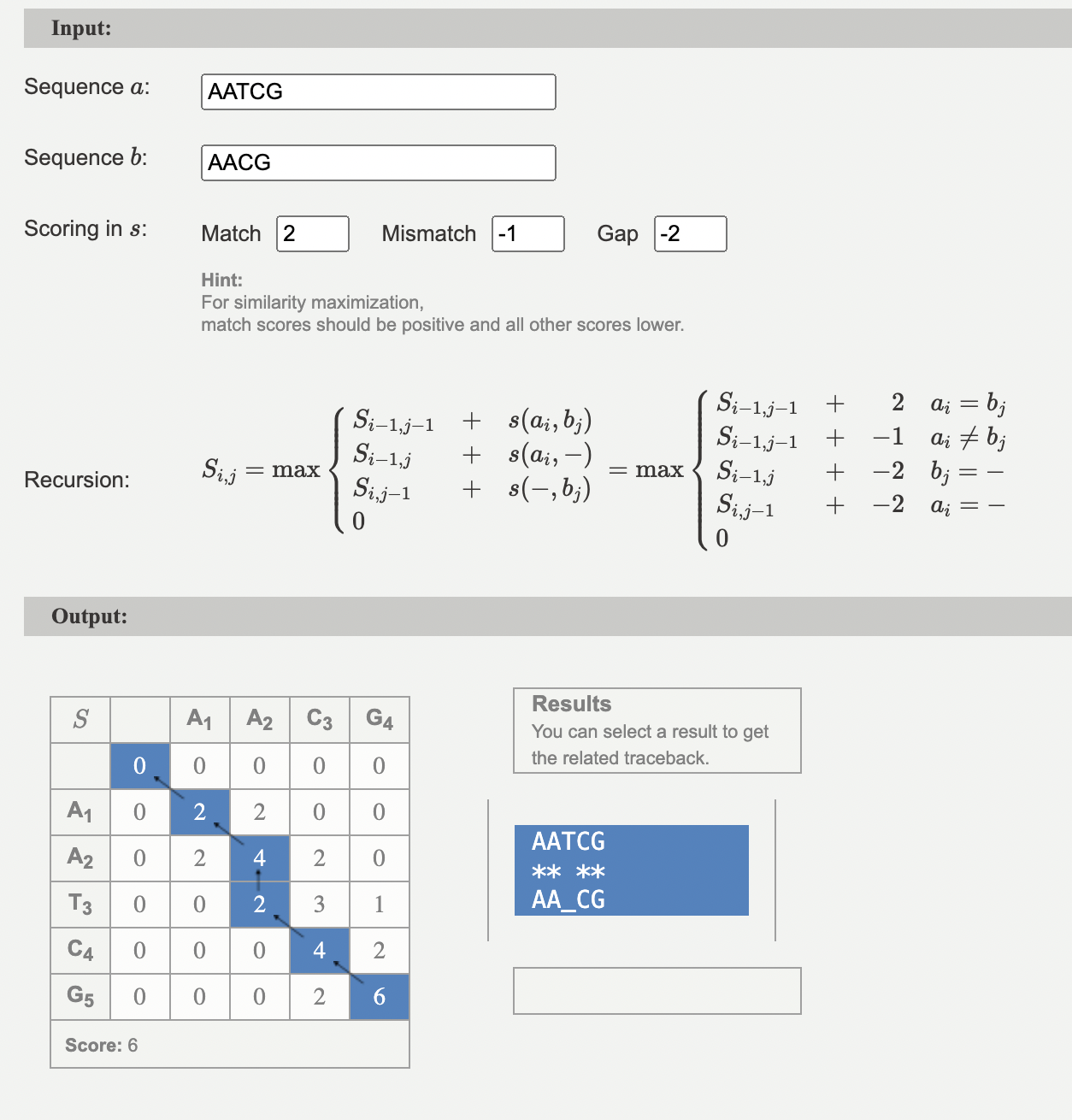
We get same output with University of Freiburg Smith-Waterman Tool (Ref 1).
Get alignment score
aligner = Align.PairwiseAligner()
aligner.mode = 'local'
alignments = aligner.align(aligned_A.replace('-',''),
aligned_B.replace('-',''))
for alignment in sorted(alignments):
print("Score = %.1f:" % alignment.score)
print(alignment)Score = 4.0:
AA-CG
||-||
AATCG